"Standard Group
俄罗斯标准集团"





医疗器械注册证和注册档案文件的修改
俄罗斯医疗器械注册证和注册档案文件的修改
Внесение изменений в регистрационное удостоверение и документы регистрационного досье медицинского изделия
俄罗斯医疗器械注册对注册档案所载文件进行无需专家审查的修改
根据俄罗斯联邦《注册规则》第 37 条,不需要对医疗器械的质量、疗效和安全性进行专家审查的变更包括
a) 申请者信息的变更,包括以下信息
法律实体重组;
法律实体名称(全称和缩写名称(如有),包括公司名称)、所在地地址的变更;
个人企业主姓氏、名字和(如有)父称、居住地址及其身份证件详细信息的变更;
b) 医疗器械注册证的签发人信息变更,包括以下信息
法律实体重组;
法人实体名称(全称和缩写名称(如有),包括公司名称)、所在地地址或姓氏、名和父称(如有)、个人企业家居住地的变更;
c) 医疗器械生产(制造)地地址变更;
d) 在影响医疗器械质量、效率和安全的特性和特征未发生变化,或其特性和特征正在改 进,而功能目的和(或)作用原理未发生变化的情况下,更改医疗器械名称:
增加(排除)医疗器械附件或更改其名称;
标明、修改和删除医疗器械的商标和其他个性化标识;
更改注册证附件中规定的医疗器械或其组成部件和组件的单位数量;
标明或排除医疗器械的变体(型号)。即不增加新版本,但在文件中提供现有版本的信息(例如,以 RU 的形式说明信息);
更改医疗器械的标签和(或)包装;
e) 医疗器械制造商(生产商)更改注册档案中文件的有效期;
f) 医疗器械制造商(生产商)授权代表信息变更。
此前(2018 年 7 月之前),不需要专家审查的注册档案修订被称为注册证书修订或缩写为 "VIRU"。因此,许多人仍在使用这一术语!
要进行不需要专家审查的修改,申请人应在相关数据发生变化之日起 30 个工作日内,按照表中的要求向俄罗斯注册局提交文件和信息:
表 1
序号 | 文件名称 Наименование документа | 备注 Примечание |
1 | 申请修改注册档案中的文件 заявление о внесении изменений в документы, содержащиеся в регистрационном досье | 应根据《登记规则》第 9 款执行 оформляется в соответствии с пунктом 9 Правил регистрации |
2 | 确认制造商(生产商)授权代表授权的文件副本 копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) | 本条规定了制造商(生产商)授权代表的职能和职责 функции и обязанности уполномоченного представителя производителя (изготовителя) приведены в этой статье |
3 | 相关变化的文件和信息 документы и сведения о соответствующих изменениях | 根据变更的性质,可能包括:租赁协议、登记簿(国家统一法人登记簿或国家统一法人登记簿)摘录等。 в зависимости от характера изменений это могут быть: договор аренды, выписки из реестров (ЕГРЮЛ или ЕГРИП) и пр. |
4 | 医疗器械名称变更时 в случае изменения наименования медицинского изделия | |
4.1 | 有关医疗器械监管文件的信息 сведения о нормативной документации на медицинское изделие | 产品符合的标准和规范性文件清单 перечень стандартов и нормативных документов, требованиям которых соответствует изделие |
4.2 | 符合新名称的医疗器械制造商(生产商)技术文件 техническая документация производителя (изготовителя) на медицинское изделие, приведенная в соответствие с его новым наименованием | 有关技术文档的更多信息,请点击此处 Подробнее о технической документации можно узнать здесь |
4.3 | 制造商(生产商)的医疗设备操作文件(包括医疗设备的使用说明或操作说明),与新名称保持一致 эксплуатационную документацию производителя (изготовителя) на медицинское изделие (в том числе инструкцию по применению или руководство по эксплуатации медицинского изделия), приведенную в соответствие с его новым наименованием | 有关操作文件的更多信息,请点击此处 Подробнее об эксплуатационной документации можно узнать здесь |
4.4 | 医疗器械一般外观的照片,以及医疗器械预期用途所需的附件(长度至少 18 厘米,宽度至少 24 厘米) фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 см в длину и 24 см в ширину) | |
5 | 制造商和(或)进行技术测试、毒理学研究、临床试验的组织的文件(相关测试结果),确认引入所申报的更改不会改变影响医疗器械质量、有效性和安全性的特性和特征,或在医疗器械的功能目的和(或)操作原理不变的情况下改进了特性和特征。документы производителя и (или) организаций, осуществляющих проведение технических испытаний, токсикологических исследований, клинических испытаний (результаты соответствующих испытаний), подтверждающие, что внесение заявленных изменений не влечет изменения свойств и характеристик, влияющих на качество, эффективность и безопасность медицинского изделия, или совершенствует свойства и характеристики при неизменности функционального назначения и (или) принципа действия медицинского изделия | 文件的构成取决于所申请变更的性质。一般来说,轻微的变更不需要提交第三方机构(如检测实验室)的文件 Состав документов зависит от характера заявленных изменений. Незначительные изменения, как правило, не требуют 第三方机构(如检测实验室)提交的材料 представления документов сторонних организаций (например, испытательных лабораторий) |
6 | 注册证书原件(一式两份) оригинал регистрационного удостоверения (дубликат) | 如果原件丢失或损坏,则提交副本 Дубликат подается в случае утери или порчи оригинала |
7 | 文件清单 опись документов | |
如果文件为外文,则必须翻译成俄文(译文必须经过认证)。
В случае если документы составлены на иностранном языке, они должны быть переведены на русский язык (перевод должен быть заверен).
进一步的操作和修改程序如图所示。
Дальнейшие действия и порядок осуществления внесения изменений приведены на схеме.
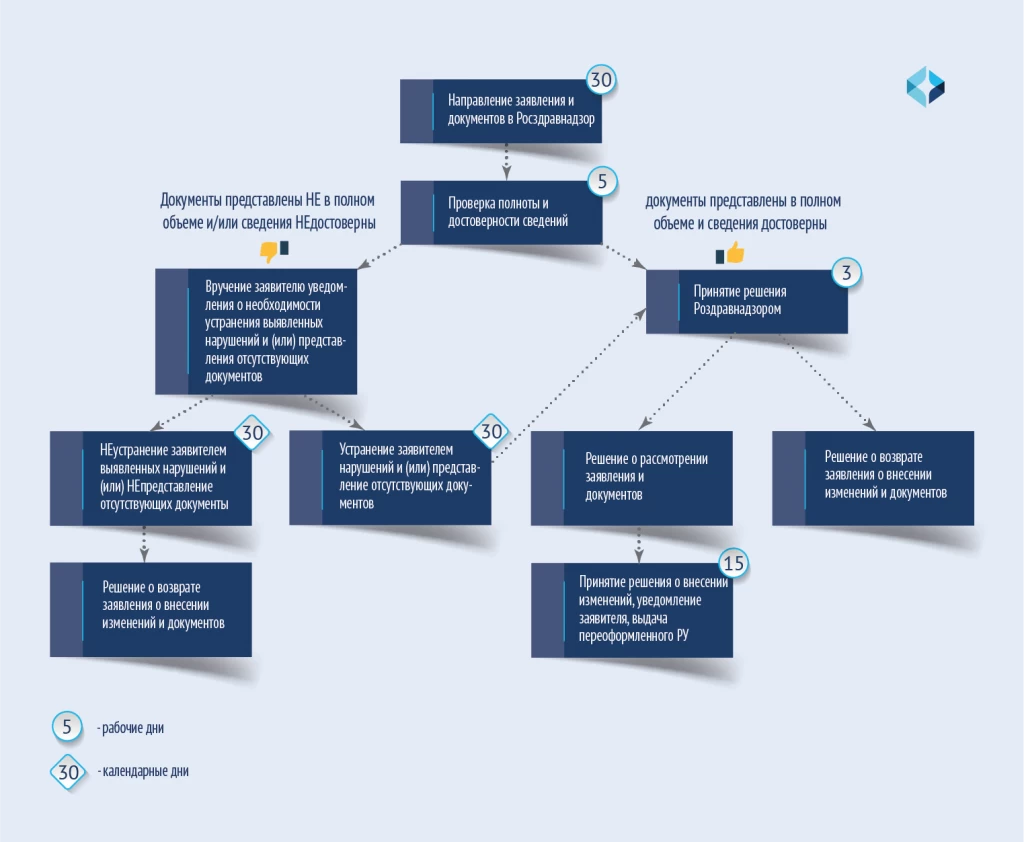
对医疗器械注册档案中的文件进行修改,如不需要对医疗器械的质量、有效性和安全性进行专家审查,需缴纳 2500 卢布的国家税。
提供修改注册证书服务的期限
在绝大多数情况下,30 个工作日足以完成不需要质量、疗效和安全专业知识的修改。
对注册档案中的文件进行修改需要专家审查
如果引入所申报的更改会导致影响医疗器械质量、疗效和安全的属性和特征发生变化,或在医疗器械的功能目的和(或)操作原理保持不变的情况下改善其属性和特征,则必须进行专家审查。
因此,在对以下方面进行更改时需要专业知识:
医疗器械制造商(生产商)的技术文件(《注册规则》第 10 段 c 分段);
医疗器械制造商(生产商)的操作文件,包括医疗器械的使用说明或操作手册(《注册规则》第 10 款 d 项)。
注册规则》第 37 条 d 款规定的变更除外:
- 在影响医疗器械质量、效率和安全的特性和特征未发生变化,或其特性和特征在功能目的和 (或)作用原理未发生变化的情况下正在改进的情况下,更改医疗器械的名称:
增加(排除)医疗器械附件或更改其名称;
标明、修改和删除医疗器械的商标和其他个性化标识;
更改注册证附件中规定的医疗器械或其组成部件和组件的单位数量;
标明或排除医疗器械的变体(型号);
改变医疗器械的标签和(或)包装;
这些变更不得影响产品的质量、疗效和安全性及其属性和特征。
更改医疗器械注册档案文件,以前简称为 "VIRD"。但在 2018 年 6 月 13 日之后,《注册规则》的修订版开始生效,将注册卷宗的变更分为需要审查的变更(原 VIRD)和不需要审查的变更(原 VIRU)!
如需对注册档案中需要专家审查医疗器械质量、疗效和安全性的文件进行变更,申请人还需在相关数据变更之日起 30 个工作日内向俄联邦药品注册局提交申请、表 1 所列文件和信息。
变更程序见下图。
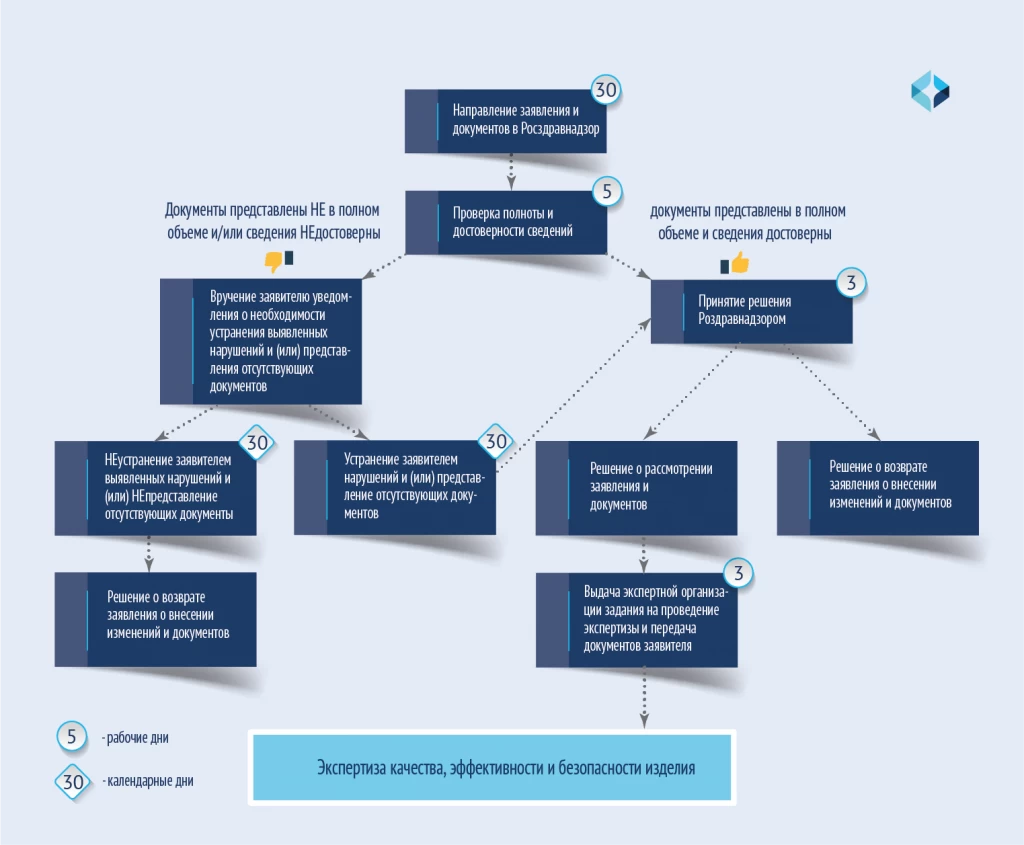
修订(VIRD)是根据专家对医疗器械质量、疗效和安全性的检查结果进行的。根据《注册规则》第 21 条进行。专家审查的步骤如下图所示。
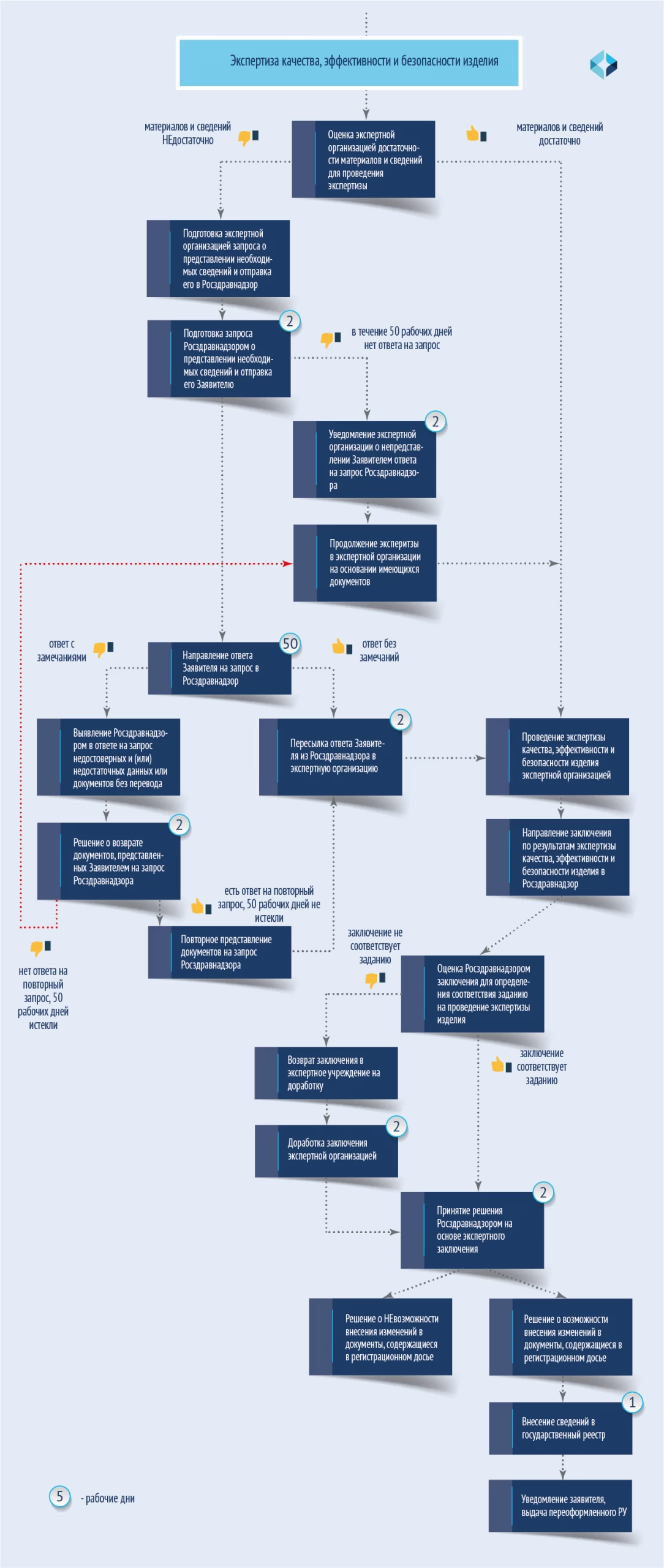
对注册档案中需要专家审查的文件进行修改的服务期限
提供服务的期限取决于许多因素,如需要更改的范围、所需测试的范围、医疗器械的风险等级、原产国等。对注册档案文件进行修改的平均期限为 3 至 6 个月。
对医疗器械的质量、疗效和安全性进行专家审查,修改注册档案文件时,需要缴纳国家税,税额取决于医疗器械的风险等级:
- 1 级 - 32000元;
- 2a 级 - 48000元
- 2b 级 - 64000元
- 3级--104000元
此外,还需支付 RI 颁发费 - 2500元。
获得登记证副本
签发登记证副本的理由包括
丢失;
损坏。
在登记证损坏的情况下,在申请登记证副本时附上损坏的登记证。
根据 2012 年 12 月 27 日第 1416 号俄罗斯联邦政府令《关于批准国家医疗器械注册规则》批准的《医疗器械注册规则》第 52 页,向俄罗斯联邦医疗器械管理局提交申请。
自收到损坏的 RI 起 7 个工作日内,Roszdravnadzor 将签发 RI 复件。在这种情况下,注册表上标有 "副本 "字样,原件上标有 "原注册证被认定无效 "字样(如果有原件的话)。然后,Roszdravnadzor 将登记证副本交给申请人(持有人),或用挂号信寄出,并要求回执。
发放登记证副本需缴纳 2500 卢布的国家税。
获得登记证副本的期限
在绝大多数情况下,12 个工作日即可获得副本。
更换登记证表格
根据俄罗斯联邦政府 2012 年 12 月 27 日第 1416 号决议第 2 段 b 分段的要求,在本决议生效日(即 2013 年 1 月 1 日之前)之前颁发的登记证可在 2021 年 1 月 1 日之前更换。
这种更换无需经过国家注册程序!目前,未更换 RI 的产品注册将被取消。因此,在国家医疗器械注册表的 "注册号有效期 "一栏中注明:"根据俄罗斯联邦政府 2012 年 12 月 27 日第 1416 号决议第 2 段 "b "项无效"。
目前,此类产品的先前注册已无法挽回地丢失,必须重新办理注册手续!
根据简化程序对医疗器械文件进行修改
随着以下法令的通过,简化修改程序成为可能:
2022 年 4 月 1 日第 552 号政府令:"关于批准医疗器械的流通特殊性,包括国家注册的特殊性,以防医疗器械存在缺陷或与俄罗斯联邦经济限制措施有关的缺陷风险";
2020 年 4 月 3 日第 430 号政府决议。"关于医疗器械流通的特殊性,包括医疗器械系列(批次)的国家注册 "的第 430 号政府决议。
让我们考虑所有可能的变革方案。
根据国家程序注册的国产医疗器械(第 552 号法令第 IV 部分)
对于已注册的国产医疗器械,2022 年 4 月 1 日第 552 号政府决议(以下简称 "决议")规定了出于以下原因更改注册档案文件的简化程序:
更改所购产品、原材料、材料和部件的信息;
更改零部件、备件和附件的数据。
这些变更需要对医疗器械的质量、有效性和安全性进行专家审查。
我们所说的是任何国产医疗器械:
- 根据《国家注册规则》(第 1416 号政府条例)注册的医疗器械,以及
- 在第 552 号法令清单中列出的和未列出的。
如需更改,应向俄联邦政府提交以下文件:
表 2
序号№ п/п | 文件名称 Наименование документа | 备注 Примечание |
1 | 申请修改文件 заявление о внесении изменений в документы | 申请必须按照《注册规则》规定的内容要求起草 заявление необходимо оформлять в соответствии с требованиями к содержанию, установленными Правилами регистрации |
2 | 确认制造商(生产商)授权代表授权的文件副本 копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) | 本条规定了制造商(生产商)授权代表的职能和职责функции и обязанности уполномоченного представителя производителя (изготовителя) приведены в этой статье |
3 | 有关更改的文件和信息,包括确认更改的文件,并证明引入这些更改不会导致医疗器械的性能和特点发生变化,影响其安全性、质量和有效性,或在保持医疗器械的功能目的和(或)操作原理不变的情况下改进其性能和特点; - 有关更改的文件和信息,包括确认更改的文件,并证明引入这些更改不会导致医疗器械的性能和特点发生变化,影响其安全性、质量和有效性,或在保持医疗器械的功能目的和(或)操作原理不变的情况下改进其性能和特点 документы и сведения о соответствующих изменениях, в том числе документы, подтверждающие изменения и свидетельствующие о том, что внесение этих изменений не влечет за собой изменения свойств и характеристик медицинского изделия, влияющих на его безопасность, качество и эффективность, или совершенствует его свойства и характеристики при неизменности функционального назначения и (или) принципа действия медицинского изделия | |
4 | 注册证书原件(一式两份) оригинал регистрационного удостоверения (дубликат) | 如果原始 RI 丢失(损坏),应提交副本 в случае утери (порчи) оригинала РУ представляется полученный дубликат |
5 | 制造商和(或)进行技术试验、毒理学研究、临床试验的组织的文件(相关试验的结果)。这些文件必须能够评估所采用的方法(技术)和所使用的测试设备清单。 这些文件必须确认,引入的变化不会导致影响产品的质量、效率和安全的特性和特征发生变化,或在功能目的和(或)操作原理不变的情况下改进了特性和特征。 документы производителя и (или) организаций, осуществляющих проведение технических испытаний, токсикологических исследований, клинических испытаний (результаты соответствующих испытаний). Документы должны позволять оценить применяемые методы (методики) и перечень используемого испытательного оборудования. документы должны подтверждать, что внесение изменений не влечет изменения свойств и характеристик, влияющих на качество, эффективность и безопасность изделия, или совершенствует свойства и характеристики при неизменности функционального назначения и (или) принципа действия | 由申请人自行决定 по выбору заявителя |
6 | 证明缴纳国家税款的文件 документы, подтверждающие оплату государственных пошлин | |
7 | 文件清单 опись документов | 清单应注明处理登记档案文件修改所依据的《命令》章节。应注明第 IV 节 в описи следует указать раздел Постановления, в соответствии с которым планируется прохождение процедуры внесения изменений в документы, содержащиеся в регистрационном досье. Следует указать раздел IV |
如果上述文件为外文,则应提交经正式公证的俄文译文。
根据决议案文,为进行上述原因的变更,无需提供变更方面的最新技术和业务文件、监管文件信息、全貌照片(与《注册规则》的要求不同)!
变更程序见该计划。
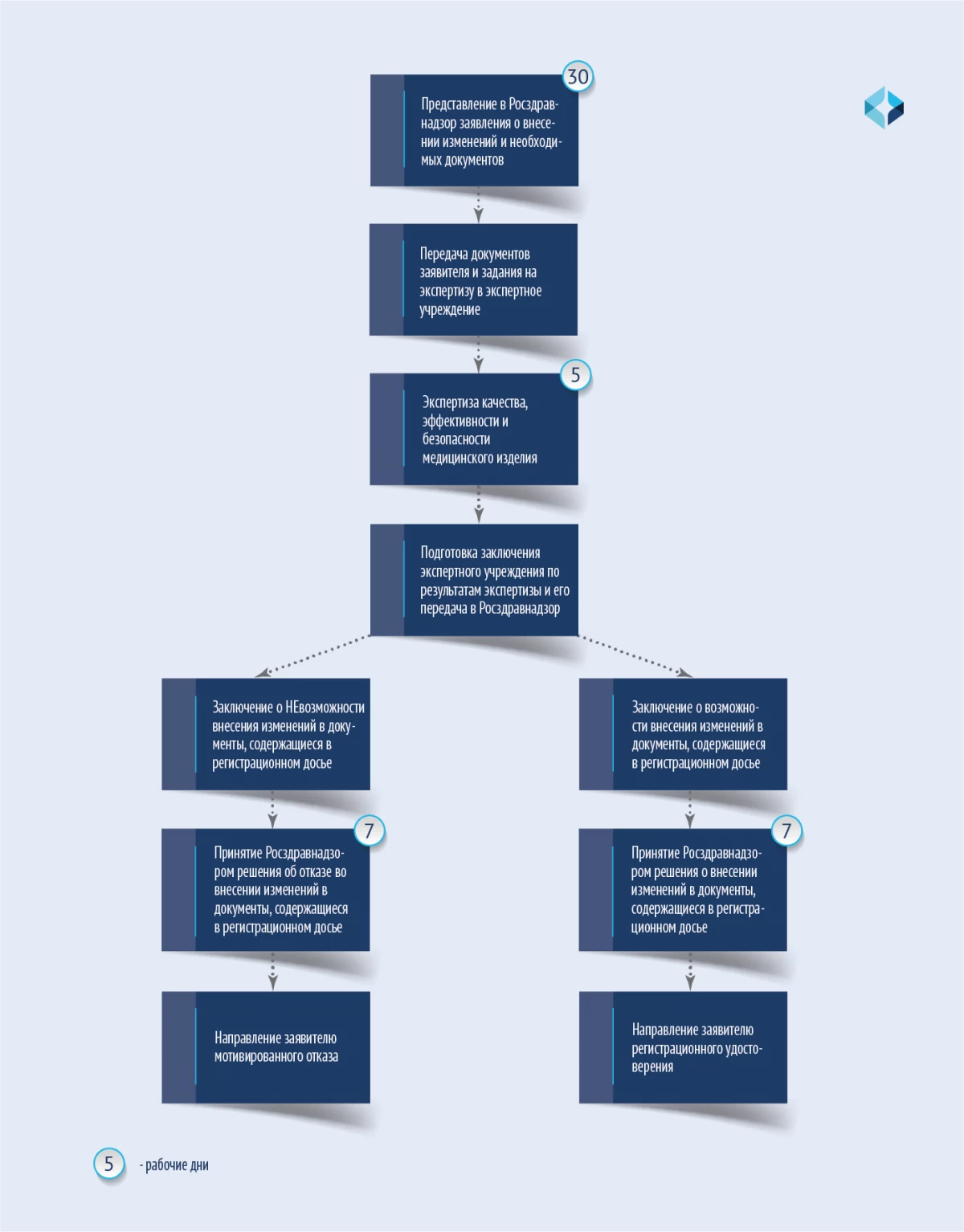
因此,质量、疗效和安全性专业知识的期限缩短为 5 个工作日,注册局作出修改注册档案或拒绝修改的决定的期限缩短为自收到结论之日起 7 个工作日。决议未规定要求提供补充材料。
第 552 号决议清单所列的风险等级为 1 的医疗器械(无菌医疗器械除外)。
对列入医疗器械清单的潜在使用风险低的医疗器械(以无菌形式生产的医疗器械除外)的注册档案中的文件进行修改,须在根据《注册规则》(见上文信息)获得无限期注册证书后进行。
第 552 号法令清单所列的所有医疗器械
无需专家审查的修改
允许根据《注册规则》规定的理由,对医疗器械注册档案中包含的文件进行更改,且无需对医疗器械的质量、功效和安全性进行专家审查,以及更改为国家注册目的将医疗器械进口到俄罗斯联邦的人员信息。
根据《注册规则》第 37 条,此类变更的理由包括
(a) 申请人信息变更,包括法人重组信息:
法律实体重组;
法律实体名称(全称和缩写(如有),包括公司名称)、所在地地址的变更;
个人企业主姓氏、名字和(如有)父称、居住地址及其身份证件详细信息的变更;
b) 医疗器械注册证的签发人信息变更,包括以下信息
法律实体重组;
法人实体名称(全称和缩写名称(如有),包括公司名称)、所在地地址或姓氏、名和父称(如有)、个人企业家居住地的变更;
c) 医疗器械生产(制造)地地址变更;
d) 在影响医疗器械质量、效率和安全的特性和特征未发生变化,或其特性和特征正在改 进,而功能目的和(或)作用原理未发生变化的情况下,更改医疗器械名称:
增加(排除)医疗器械附件或更改其名称;
标明、修改和删除医疗器械的商标和其他个性化标识;
更改注册证附件中规定的医疗器械或其组成部件和组件的单位数量;
标明或排除医疗器械的变体(型号);
改变医疗器械的标签和(或)包装;
e) 医疗器械制造商(生产商)更改注册档案中文件的有效期;
f) 医疗器械制造商(生产商)授权代表的信息变更;
根据 2022 年 4 月 1 日第 552 号决议第 18 条规定的理由:为进行国家注册而将医疗器械进口到俄 罗斯联邦的人员信息变更。
如需更改,申请人应直接向俄罗斯注册局提交以下文件的硬拷贝,或通过挂号信寄送,并要求回执和清单:
表 3
序号№ п/п | 文件名称Наименование документа | 备注 Примечание |
1 | 申请修改文件 заявление о внесении изменений в документы | 申请必须按照《注册规则》对修改申请内容的要求执行 заявление должно быть оформлено в соответствии с требованиями к содержанию заявления о внесении изменений, установленными Правилами регистрации |
2 | 确认制造商(生产商)授权代表授权的文件副本 копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) | 本条规定了制造商(生产商)授权代表的职能和职责 функции и обязанности уполномоченного представителя производителя (изготовителя) приведены в этой статье |
3 | 医疗器械进口到俄罗斯联邦进行国家注册的授权确认文件原件 оригинал документа, подтверждающего полномочия лица, осуществляющего ввоз медицинского изделия в Российскую Федерацию в целях его государственной регистрации | |
4 | 证明该医疗器械属于俄罗斯联邦进口医疗器械者的文件,以便根据法律规定进行国家注册 документы, подтверждающие принадлежность медицинского изделия лицу, осуществляющему ввоз медицинского изделия в Российскую Федерацию в целях его государственной регистрации на законных основаниях | |
5 | 证明可以在修改申请中指定的地址(生产基地)进行生产的文件 документы, подтверждающие возможность осуществления производства по адресу (адресам), указанному (указанным) в заявлении о внесении изменений (производственная площадка (производственные площадки) | 确认生产条件可用性的文件,以及(或)质量管理体系符合 GOST ISO 13485 或相关国际标准 ISO 13485 要求的证书副本(如果有的话) документы, подтверждающие наличие условий производства, и (или) копии сертификатов соответствия системы менеджмента качества требованиям стандарта ГОСТ ISO 13485 или соответствующего международного стандарта ISO 13485) (при наличии) |
6 | 有关更改的文件和信息,包括确认更改的文件,并证明引入这些更改不会导致医疗器械的性能和特点发生变化,影响其安全性、质量和有效性,或在保持医疗器械的功能目的和(或)操作原理不变的情况下改进其性能和特点; - 有关更改的文件和信息,包括确认更改的文件,并证明引入这些更改不会导致医疗器械的性能和特点发生变化,影响其安全性、质量和有效性,或在保持医疗器械的功能目的和(或)操作原理不变的情况下改进其性能和特点 документы и сведения о соответствующих изменениях, в том числе документы, подтверждающие изменения и свидетельствующие о том, что внесение этих изменений не влечет за собой изменения свойств и характеристик медицинского изделия, влияющих на его безопасность, качество и эффективность, или совершенствует его свойства и характеристики при неизменности функционального назначения и (или) принципа действия медицинского изделия | 检测实验室或制造商自己进行的检查和测试的结果 результаты исследований и испытаний, выполненные в испытательных лабораториях или самим производителем |
7 | 注册证书原件(一式两份) оригинал регистрационного удостоверения (дубликат) | 如果原始 RI 丢失(损坏),应提交副本 в случае утери (порчи) оригинала РУ представляется полученный дубликат |
8 | 注册机构为进口医疗器械进行国家注册而颁发的许可证信息。对于其制造商为在俄罗斯联邦境内注册的法人实体或个体工商户,且该医疗器械的制造地(生产基地)地址位于外国境内的产品,以及外国制造的医疗器械,但不包括属于医疗器械的软件,包括使用人工智能技术的软件。 сведения о выданных регистрирующим органом разрешениях на ввоз медицинских изделий с целью их государственной регистрации. Для изделий, производителем которых является юридическое лицо или индивидуальный предприниматель, зарегистрированный на территории РФ, а адрес (адреса) места (мест) производства (производственная площадка (производственные площадки) таких медицинских изделий находится (находятся) на территории иностранного государства, и медицинских изделий иностранного производства, за исключением ПО, являющегося медицинским изделием, в том числе ПО с применением технологий искусственного интеллекта | |
9 | 文件清单 опись документов | 清单应注明处理登记档案文件修改所依据的命令章节。应注明第二节 в описи следует указать раздел Постановления, в соответствии с которым планируется прохождение процедуры внесения изменений в документы, содержащиеся в регистрационном досье. Следует указать раздел II |
10 | 证明缴纳国家税款的文件 документы, подтверждающие оплату государственных пошлин |
如果上述文件为外文,则应提交经正式认证的俄文译文。
制造商(生产商)的文件,除 PSO 的委托书外,应由制造商(生产商)或授权代表进行认证,但必须提供一份文件,确认 PSO 在执行表中所列文件之前有权对其进行认证和生效,并将其提交给俄罗斯国家质量监督检验检疫总局。
根据该法令文本,由于上述原因,无需提供技术和运行文件、监管文件信息、全景照片(与《注册规则》的要求不同)的更新变更!
根据本方案进行的修改。
第 552 号法令第 2 条规定的变更方案
需要专家审查的修改
对根据本法令注册的医疗器械的注册档案文件的修改,如需要对医疗器械的质量、有效性和安全性进行专家审查,应按照《注册规则》规定的程序进行。此类修改应在收到修改申请和《注册规则》规定的整套文件之日起不超过 25 个工作日的期限内完成。修改的标准期限为 35 个工作日。
根据第 430 号法令进行修改
对于根据第 430 号法令注册并在该法令附件 1 第 1 至 18 段中指明的医疗器械,可对以下内容进行修改:
医疗器械名称,更改其出厂编号信息(如有);
系列(批次)编号。
同时,不得更改注册档案文件中的其他信息。
如需更改,申请人必须向俄罗斯注册局提交以下文件:
表 4
序号№ п/п | 文件名称 Наименование документа | 备注 Примечание |
1 | 申请修改注册档案中的文件 заявление о внесении изменений в документы, содержащиеся в регистрационном досье | 应根据《登记规则》第 9 条执行 оформляется в соответствии с п.9 Правил регистрации |
2 | 确认制造商(生产商)授权代表授权的文件副本 копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) | 如果有的话。制造商(生产商)授权代表的职能和职责见本条。 при наличии. Функции и обязанности уполномоченного представителя производителя (изготовителя) приведены в этой статье |
3 | 根据法律依据确认该系列(批次)医疗器械属于申请人的文件документы, подтверждающие принадлежность серии (партии) медицинского изделия заявителю на законных основаниях | |
4 | 医疗器械一般外观的照片,以及医疗器械预期用途所需的附件(尺寸不小于 18×24 厘米) фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для применения медицинского изделия по назначению (размером не менее 18 на 24 сантиметра) | |
5 | 制造商的文件,确认带有申报出厂编号(如有)、系列(批次)编号的医疗器械与根据本文件进行初始注册的医疗器械完全相同 документ производителя, подтверждающий, что медицинское изделие с заявляемыми заводскими номерами (при наличии), номерами серии (партии) идентично медицинскому изделию, зарегистрированному первоначально в соответствии с настоящим документом | |
6 | 原始注册证书 оригинал регистрационного удостоверения | 此 RI 更改为包含新信息的重新发布的 RI данное РУ меняется на переоформленное с новыми сведениями |
7 | 文件清单 опись документов |
如果上述文件为外文,则应提交经申请人认证的俄文译文。
В случае если указанные документы составлены на иностранном языке, они представляются с заверенным заявителем переводом на русский язык.
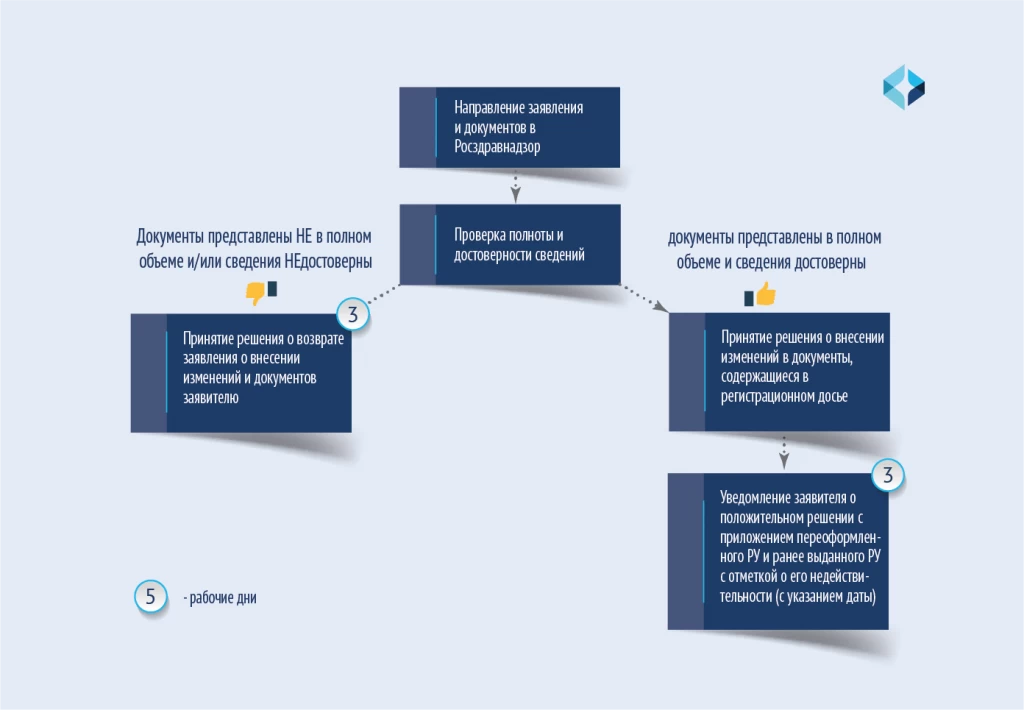
根据第 430 号法令进行更改的步骤如图所示。
Действия по внесению изменений в соответствии с постановлением №430 приведены на схеме.
因此,修改医疗器械注册档案文件和注册证书的选择相当多。如果您在医疗器械流通方面有任何疑问,我们的专家将为您提供免费咨询,并在任何情况下为您找到最佳解决方案。请通过您方便的方式联系我们!
海关联盟EAC认证中心/上海经合工业设备检测有限公司
公司地址:上海浦东新区高科东路777弄1号楼2017室
联系我们:上海经合工业设备检测有限公司/俄罗斯EAC证机构中国代表处
电话:021-36411223 36411293
邮件:eac@cu-tr.org
手机微信:18621862553

欧亚联盟EAEU、俄罗斯、乌兹别克斯/哈萨克斯坦/白俄罗斯/乌克兰医疗器械和药品注册咨询服务
Eurasian Economic Union Russia Uzbekistan Kazakhstan Belarus Ukraine Medical Device and Drug Registration Consulting Services
Moscow, st. Lenin Sloboda, d.21, k.1 浙江省杭州市西湖区留和路129号2924室
China Office: Room 2017, Building 1, No. 777, Gaoke East Road, Pudong New Area, Shanghai,china
微信/电话:18621327282 QQ:594037022 gost-r@163.com 邮编:210201 海关编码查询 俄罗斯医疗器械注册证查询 俄罗斯医疗注册证书查询
网站内容和图片版权所有,未经允许,不得转载复制拷贝和使用 沪ICP备16045803号-6 沪ICP备10027014号-18 欧亚联盟药品注册证查询 俄罗斯计量注册证书查询
