"Standard Group
俄罗斯标准集团"




欧亚联盟和俄罗斯医疗器械临床试验要求
俄罗斯医疗器械临床试验要求
Клинические испытания медицинских изделий
俄罗斯医疗器械的临床试验是医疗器械成功注册所需的一种强制性测试。临床试验确认医疗器械在医疗实践中使用的可能性。
Клинические испытания медицинских изделий – это обязательный вид испытаний, необходимых для их успешной регистрации. Именно они подтверждают возможность использования медицинского изделия в медицинской практике.
俄罗斯临床试验是在收到技术和毒理学测试结果后进行的,对监管机构决定医疗器械是否注册起着决定性作用。
Клинические испытания проводятся после получения результатов технических и токсикологических испытаний и являются решающими для принятия регулятором решения о регистрации медицинского изделия.
如果临床试验结果不令人满意,在大多数情况下,必须从头开始注册。这意味着制造商在注册过程中需要花费大量的额外时间和成本。
В случае получения неудовлетворительных результатов клинических испытаний в большинстве случаев потребуется начинать регистрацию с самого начала. Это сулит для производителей значительные дополнительные затраты времени и средств на регистрацию.
开展形式
Формы проведения
根据医疗器械的类型和现有的临床数据,临床试验可以以下列形式进行:
Клиника может быть проведена в следующих формах, в зависимости от вида медицинского изделия и имеющихся клинических данных:
1、研究形式(分析和评估现有数据);
в форме исследований (анализа и оценки доступных данных);
2、人体试验
в форме испытаний с участием человека;
3、临床和实验室试验(体外诊断产品)。
в форме клинико-лабораторных испытаний (изделия для диагностики in vitro).
分析和评估现有数据形式的临床试验
Клинические испытания в форме анализа и оценки доступных данных
调查形式的试验是最简单快捷的方法。目前俄罗斯联邦绝大多数临床试验都是以这种形式进行的。为此,只需收集医疗器械的临床数据(通常适用于已在其他国家上市的外国产品)或先前在俄 罗斯联邦注册的类似产品的临床数据(适用于国产产品)即可。
Испытания в форме исследований − самый простой и быстрый способ. Подавляющее большинство клинических испытаний в РФ в настоящее время проводится именно в этой форме. Для этого достаточно собрать клинические данные либо по медицинскому изделию (как правило, для зарубежных изделий, которые уже обращаются на рынках других стран), либо по ранее зарегистрированному в РФ аналогу (для изделий отечественного производства).
根据现有的临床数据,对注册产品的质量、安全性和有效性进行评估和分析。然后,对其在医疗实践中使用的可能性做出结论。
На основании имеющихся клинических данных проводится оценка и анализ качества, безопасности и эффективности регистрируемого изделия. После чего делается вывод о возможности его использования в медицинской практике.
人体试验形式的临床试验
Клинические испытания в форме испытаний с участием человека
这是最昂贵、最耗时的方法。它需要大量的资金和时间来进行。在某些情况下,如植入试验,由于缺乏符合纳入标准的患者,时间可能会延长数年。
Это самый дорогой и длительный способ. Требует значительного количества средств и времени на проведение. В некоторых случаях, например, при испытаниях имплантатов, сроки могут растянуться на годы ввиду отсутствия пациентов, удовлетворяющих критериям включения в исследования.
这种形式的临床试验需要获得俄罗斯国家科研署(Roszdravnadzor)的授权!
Именно для этой формы требуется получение разрешения на проведение клинических испытаний в Росздравнадзоре!
不过,目前绝大多数情况下都不需要进行有人体参与的试验。
Однако, в настоящее время необходимость проведения испытаний с участием человека в подавляющем большинстве случаев отсутствует.
需要进行人体临床试验时
В каких случаях необходимы клинические испытания с участием человека
俄罗斯联邦卫生部2021年8月30日第885n号 "关于批准以技术试验、毒理学研究、临床试验 形式对医疗器械进行符合性评估以实现医疗器械国家注册 "的命令(以下简称第 885n 号命令)第37条规定了以下需要人体参与试验的情况:
п. 37 Приказа Министерства Здравоохранения РФ от 30 августа 2021 г. N 885н «Об утверждении порядка проведения оценки соответствия медицинских изделий в форме технических испытаний, токсикологических исследований, клинических испытаний в целях государственной регистрации медицинских изделий» (далее ‒ Приказ 885н), устанавливает следующие случаи, требующие выполнения испытаний с участием человека:
医疗器械注册属于术语分类中的新类型;
регистрируемое медицинское изделие относится к новому виду в соответствии с номенклатурной классификацией;
新的复杂和/或独特和/或特殊的预防、诊断和治疗疾病和病症的方法,以及新的复杂医疗技术已应用于注册医疗器械;
в регистрируемом медицинском изделии применены новые сложные и/или уникальные и/или специальные методы профилактики, диагностики и лечения заболеваний и состояний равно как применены новые сложные медицинские технологии;
注册的医疗器械在生产过程中使用了与人体接触的新材料,而这些材料的生物效应以前未曾研究过,或使用了与人体器官或组织接触的已知材料,而这些材料在医疗上的使用还没有经验,或这种接触的时间比以前研究过的要长;
изделие, подлежащее регистрации, изготовлено с использованием новых, контактирующих с организмом человека, ранее не изученных в части биологического действия материалов или известных материалов, контактирующих с теми органами или тканями человека, в отношении которых отсутствует опыт их медицинского применения, или в случае, если такой контакт является более продолжительным, чем ранее изученный;
医疗器械的有效性和安全性未经临床数据分析和评估确认
если при проведении анализа и оценки клинических данных не были подтверждены эффективность и безопасность медицинского изделия.
在所有其他情况下,诊所以分析和评估现有临床数据的形式进行。
Во всех остальных случаях клиника проводится в форме анализа и оценки доступных клинических данных.
临床和实验室试验(体外诊断产品)
Клиника в форме клинико-лабораторных испытаний
这种形式是为了体外诊断医疗器械的注册而进行的。对于这类医疗器械,现行法律只规定了一种试验方式。
В этой форме проводится для целей регистрации медицинских изделий для in vitro диагностики. Для этого вида медицинских изделий действующим законодательством предусмотрен только один способ проведения испытаний.
您可以在本页面阅读有关 IVD 医疗器械临床试验的更多信息。
Подробнее о клинических испытаниях медицинских изделий для ИВД можно прочесть на это странице.
临床试验(非体外)所需文件
Документы, необходимые для проведения клинических испытаний (не in vitro)
根据第 885n 号令第 38 条,医疗器械临床试验(体外诊断器械除外)必须向医疗组织提交以下文件:
Согласно п.38 Приказа 885н, для проведения клинических испытаний медицинских изделий (за исключением изделий для диагностики in vitro) необходимо представить в медицинскую организацию следующие документы:
序号№п/п | 文件名称 Наименование документа | 说明 Комментарии |
1 | 临床试验申请 Заявление о проведении клинических испытаний | 申请应提交给符合俄罗斯卫生部 2013 年 5 月 16 日第 300n 号命令要求的医疗机构 Заявление подается в медицинскую организацию, соответствующую требованиям Приказа Минздрава России от 16.05.2013 № 300н |
2 | 医疗器械样品(样本) Образцы (образец) медицинского изделия | 但医疗器械除外,其安装(调试)需要获得许可(执照)、创造特殊条件、建造独立的资本结构和对专家进行额外培训,在某些情况下,还需要离开医疗器械放置地的组织,并(或)根据离开国的法律允许使用。) 在这种情况下,应在产品放置地进行试验。 За исключением медицинских изделий, для монтажа (ввода в эксплуатацию) которых требуется получение разрешений (лицензий), создание специальных условий, строительства отдельных капитальных сооружений и дополнительного обучения специалистов, а в некоторых случаях — выезд в организацию, где медицинское изделие размещено и (или) разрешено для применения в соответствии с законодательством страны, в которую осуществляется выезд). В таких случаях испытания проводят на месте размещения изделий. |
3 | 俄罗斯国家科研署颁发的临床试验许可证 Разрешение на проведение клинических испытаний, выданное Росздравнадзором | 只有在进行有人类参与的试验形式的诊所时才需要授权 Разрешение нужно только при проведении клиники в форме испытаний с участием человека |
4 | 医疗器械技术检测结果评估法,附技术检测结果证明文件 Акт оценки результатов технических испытаний медицинского изделия с приложением документов, обосновывающих результаты технических испытаний | 关于技术测试的更多信息 Подробнее о о технических испытаниях |
5 | 关于医疗器械毒理学研究结果的结论,并附上证明毒理学研究结果的文件 Заключение по результатам токсикологических исследований медицинского изделия с приложением документов, обосновывающих результаты токсикологических исследований | 用于直接或间接接触人体(人体表面、粘膜、内部环境)的物品和/或配件。通常不对体外产品进行毒理学研究。毒理学研究的更多信息 Для изделий и/или принадлежностей, использование которых предполагает наличие прямого или опосредованного контакта с организмом человека (поверхностью тела человека, его слизистыми оболочками, внутренними средами организма). Как правило не проводятся для изделий in vitro. Подробнее о токсикологических исследованиях |
6 | 测量仪器型式批准的试验结果 Результаты испытания в целях утверждения типа средств измерений | 在保证测量统一性的国家规定范围内,与测量仪器有关的项目。这些产品的清单由俄罗斯联邦卫生部批准。有关型式批准试验的更多信息 Для изделий, относящихся к средствам измерений в сфере государственного регулирования обеспечения единства измерений. Перечень этих изделий утверждается Министерством здравоохранения Российской Федерации. Подробнее об испытаниях в целях утверждения типа |
7 | 医疗器械的监管文件信息,包括符合的国家(国际)标准清单 Сведения о нормативной документации на медицинское изделие с перечнем национальных (международных) стандартов, требованиям которых оно соответствует | |
8 | 医疗设备制造商的技术和操作文件 Техническая и эксплуатационная документация производителя на медицинское изделие | 了解更多技术文档信息 . 了解更多操作文档 Подробнее о технической документации . Подробнее об эксплуатационной документации |
9 | 医疗器械一般外观的彩色照片,以及预期用途所需的附件 Цветные фотографические изображения общего вида медицинского изделия вместе с принадлежностями, необходимыми для его применения по назначению | 尺寸至少为 18 x 24 厘米。所有版本的设计都必须提交图片 Размером не менее 18 x 24 см. Изображения должны быть представлены для всех вариантов исполнения |
10 | 电子媒体和软件界面的彩色摄影图像 Цветные фотографические изображения электронного носителя и интерфейса программного обеспечения | 尺寸至少为 18 x 24 厘米。所有版本的设计都必须提交图片 Размером не менее 18 x 24 сантиметра. Только для программного обеспечения, являющегося медицинским изделием |
11 | 可下载医疗器械软件的互联网资源链接(如果软件没有电子媒介),以及医疗器械制造商(生产商)提供的访问密钥和密码列表。 Ссылка на ресурс в сети интернет, с которого может быть загружено ПО, являющееся медицинским изделием (в случае отсутствия у ПО электронного носителя), а также перечень предоставленных производителем (изготовителем) медицинского изделия ключей, паролей доступа | 如有 При наличии |
12 | 标签和包装数据(包含俄文标识文字的包装和标签彩色布局图) Данные о маркировке и упаковке (цветные макеты упаковок и этикеток, содержащие текст маркировки на русском языке) | 属于医疗设备的软件除外,包括使用人工智能技术的软件 За исключением программного обеспечения, являющегося медицинским изделием, в том числе ПО с применением технологий ИИ |
13 | 包含医疗器械临床使用数据的文件(资料),包括俄罗斯境外的文件(资料)。包括综述、科学研究报告、出版物、报告、使用风险分析、使用方法 Документы (материалы), содержащие данные о клиническом применении медицинского изделия, в том числе за пределами России. В том числе обзоры, отчеты о проведенных научных исследованиях, публикации, доклады, анализ риска применения, методы применения | 如有 При наличии |
14 | 关于俄罗斯国家工业技术监督局(Roszdravnadzor)颁发的 MI 进口许可证的信息,包括为修改登记档案中的文件而颁发的许可证的信息 Сведения о выданных Росздравнадзором разрешениях на ввоз МИ, в том числе в целях внесения изменений в документы, содержащиеся в регистрационном досье | 对于外国制造的产品,以及进口到俄罗斯境内的 MI,其制造商是俄罗斯联邦居民的法人实体或个体企业家,其生产地位于外国境内。但属于医疗器械的软件,以及俄罗斯联邦政府于 2012 年 12 月 27 日批准的《使用潜在风险较低的医疗器械清单》中所列的器械除外。 Для изделий иностранного производства, а также в случае ввоза на территорию России МИ, производителем которого является юридическое лицо или индивидуальный предприниматель - резиденты РФ, а место его производства находится на территории иностранного государства. За исключением ПО, являющегося медицинским изделием, и изделий, включенных в перечень медицинских изделий с низкой степенью потенциального риска их применения, в отношении которых установлены особенности государственной регистрации, утвержденный постановлением Правительства Российской Федерации от 27 декабря 2012 г. N 1416 |
15 | 关于注册档案变更的文件和信息,包括确认变更的文件 Документы и сведения об изменениях, вносимых в регистрационное досье, в том числе документы, подтверждающие данные изменения | 为修改注册档案中的文件而对医疗器械进行检测时 При проведении испытаний медицинского изделия в целях внесения изменений в документы, содержащиеся в регистрационном досье |
16 | 确认制造商(生产商)授权代表授权的文件副本 Копия документа, подтверждающего полномочия уполномоченного представителя производителя (изготовителя) | 对于外国制造的产品。有关制造商授权代表的更多信息 Для изделий иностранного производства. Подробнее об уполномоченном представителе производителя |
如果原始文件为外文,则需提交经认证的俄文译文。
В случае если оригиналы документов составлены на иностранном языке, они представляются с заверенным переводом на русский язык.
体外诊断产品临床试验所需文件列于网站本部分的表格中。
Документы, необходимые для проведения клинических испытаний изделий для in vitro диагностики, приведены в таблице этого раздела сайта.
进行医疗器械临床试验的程序
Порядок проведения клинических испытаний медицинских изделий
进行试验的详细步骤以弹出提示的方式呈现。将鼠标悬停在这些步骤上时,窗口会显示申请人必须考虑的要求、每个阶段的内容以及需要特别注意的要点。
Детальные действия по проведению испытаний представлены на схеме с всплывающими подсказками. При наведении на них в окне отражаются требования, которые должен учесть Заявитель, содержание каждого из этапов и важные моменты, на которые стоит обращать особое внимание.
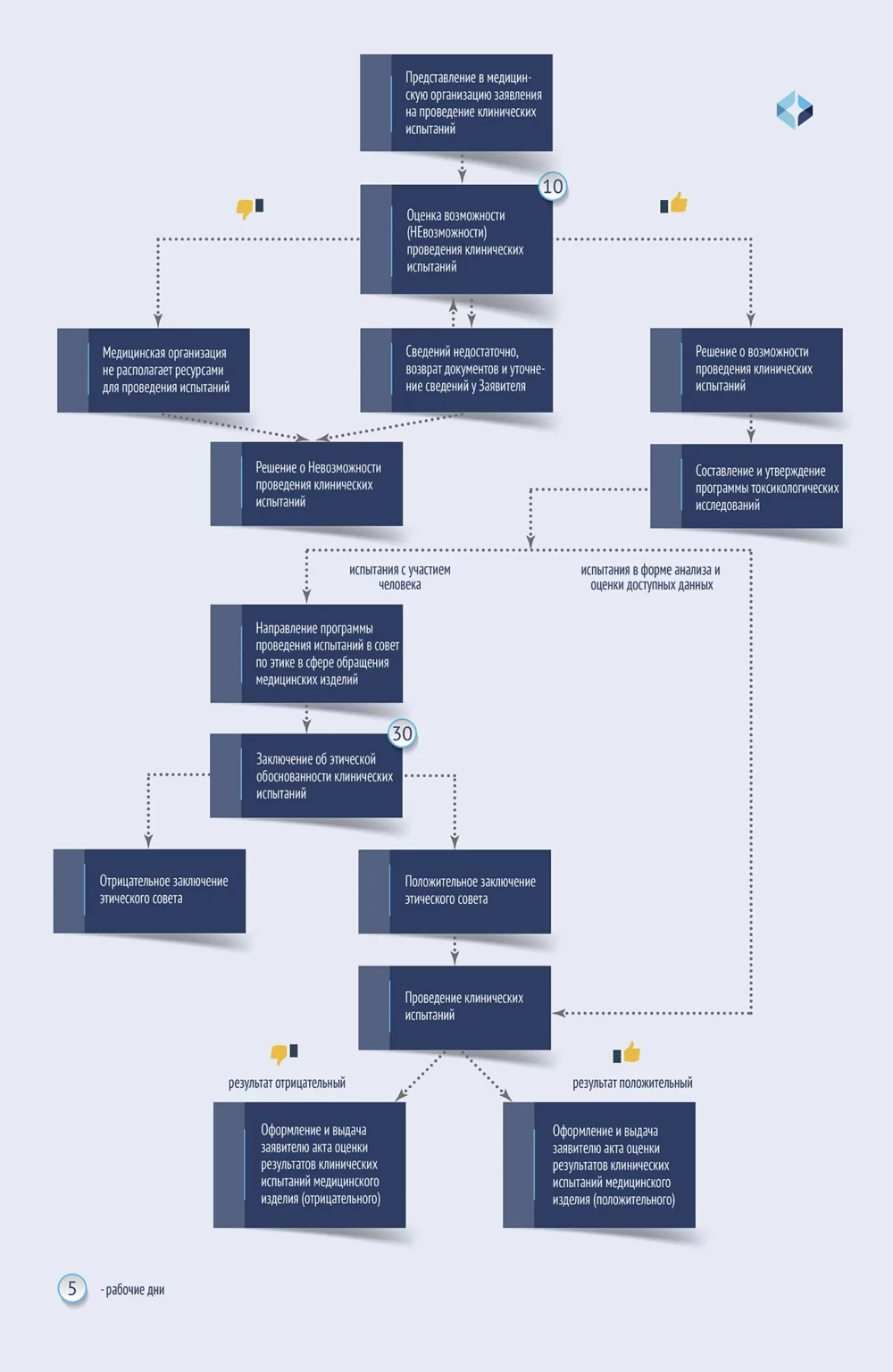
根据结果,制定医疗器械临床试验结果评估法案。
По результатам оформляется акт оценки результатов клинических испытаний медицинского изделия.
有关体外诊断医疗器械临床试验的具体内容,请参阅本页。
Особенности клинических испытаний медицинских изделий для диагностики in vitro детально рассмотрены на этой странице.
临床试验的内容
Что включают в себя клинические испытания
在进行临床试验时,需要开展以下工作:
При их проведении проводится:
分析和评估申请人提交的临床数据、文件和材料
анализ и оценка клинических данных, документов и материалов, представленных Заявителем;
评估有关具有临床意义的纠正措施的信息,包括暂停使用医疗器械、从流通中撤回医疗器械、医疗器械召回
оценка сведений о проводившихся клинически значимых корректирующих действиях, в том числе о приостановлении применения медицинского изделия, об изъятии из обращения медицинского изделия, об отзывах медицинского изделия;
分析科学文献和(或)未发表的数据和报告,这些数据和报告涉及制造商对受 检医疗器械的预期用途和建议的使用方法;
анализ научной литературы и (или) неопубликованных данных и сообщений, соотнесенных с предназначенным производителем применением испытуемого медицинского изделия и предлагаемым методом его использования;
在人体试验中对医疗器械样品进行测试;
проведение испытаний образца (образцов) медицинского изделия в случае проведения испытаний с участием человека;
评估作为医疗器械的软件(包括应用人工智能技术的软件)的临床通信、分析和临床验证的可靠性;
оценка достоверности клинической связи, аналитической и клинической валидации программного обеспечения, являющегося медицинским изделием, в том числе программного обеспечения с применением технологий искусственного интеллекта;
根据测试结果(如有必要),最终确定医疗器械制造商(生产商)的操作文件;
доработка эксплуатационной документации производителя (изготовителя) на медицинское изделие по результатам испытаний (при необходимости);
分析和评估有关可互换医疗器械(在功能用途、质量和技术特性方面具有可比性并可相互替代的医疗器械)的信息;
анализ и оценка сведений о взаимозаменяемых медицинских изделиях (медицинских изделиях сравнимых по функциональному назначению, качественным и техническим характеристикам и которые способны заменить друг друга);
起草医疗器械临床试验结果评估报告并向申请人签发(签字后亲自递交或挂号邮寄并附上递交通知)。
оформление и выдача (вручение лично под подпись или направление заказным почтовым отправлением с уведомлением о вручении) заявителю акта оценки результатов клинических испытаний медицинского изделия.
在临床试验中确定什么?
Что определяется в ходе клинических испытаний?
在试验过程中要确定
В ходе проведения испытаний определяются:
医疗器械是否符合监管文件、制造商(生产商)的技术和操作文件;
соответствие медицинского изделия нормативной документации, технической и эксплуатационной документации производителя (изготовителя);
申请人提交的文件是否符合制造商的处方和使用说明;
соответствие представленной заявителем документации установленным производителем назначению и показаниям к применению;
生产商的监管文件、技术和操作文件所确定的医疗器械特性的完整性和可靠性;
полнота и достоверность установленных нормативной документацией, технической и эксплуатационной документацией производителя характеристик медицинского изделия;
医疗产品的质量、使用的有效性和安全性。
качество медицинского изделия, эффективность и безопасность его применения.
根据第 885n 号令第 46 条,临床试验结果为不符合的情况:
Случаи отрицательного результата клинических испытаний в соответствии с п.46 Приказа 885н:
医疗器械不符合制造商在医疗器械操作文件中规定的使用目的和适应症;
медицинское изделие не соответствует назначению и показаниям к применению, установленным производителем в эксплуатационной документации на медицинское изделие;
在医疗器械的使用和操作过程中,对公民和医务工作者的生命和健康构成威胁的事实和情况已经确定;
установлены факты и обстоятельства, создающие угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинского изделия;
在医疗器械的使用说明或操作手册中未明确说明的副作用,以及在使用过程中发现的不良反应。
выявлены побочные действия, не указанные в инструкции по применению или руководстве по эксплуатации медицинского изделия, нежелательные реакции при его применении.
在所有其他情况下,临床试验结果均被视为阳性(合格)。这意味着医疗器械在按照制造商(生产商)文件规定的预期用途使用时是安全有效的。
Во всех остальных случаях результаты клинических испытаний считаются положительными. Это означает, что медицинское изделие безопасно и эффективно при его применении в соответствии с назначением, предусмотренным документацией производителя (изготовителя).
可进行临床试验的地点
Где могут быть проведены клинические испытания
只有符合俄罗斯卫生部2013年5月16日第300n号 "关于批准医疗机构进行医疗器械临床试验的要求和确定医疗机构符合这些要求的程序 "命令要求的医疗机构才能进行试验。
Испытания могут быть проведены только в медицинских учреждениях, отвечающих требованиям Приказа Минздрава России от 16.05.2013 № 300н «Об утверждении требований к медицинским организациям, проводящим клинические испытания медицинских изделий, и порядка установления соответствия медицинских организаций этим требованиям».
符合该命令要求的组织的最新名单可在联邦医疗保健监督局的网站上查阅(见下图)。
Актуальный перечень организаций соответствующих требованиям данного приказа размещен на сайте Федеральной службы по надзору в сфере здравоохранения (см. рисунок Сервиса ниже).
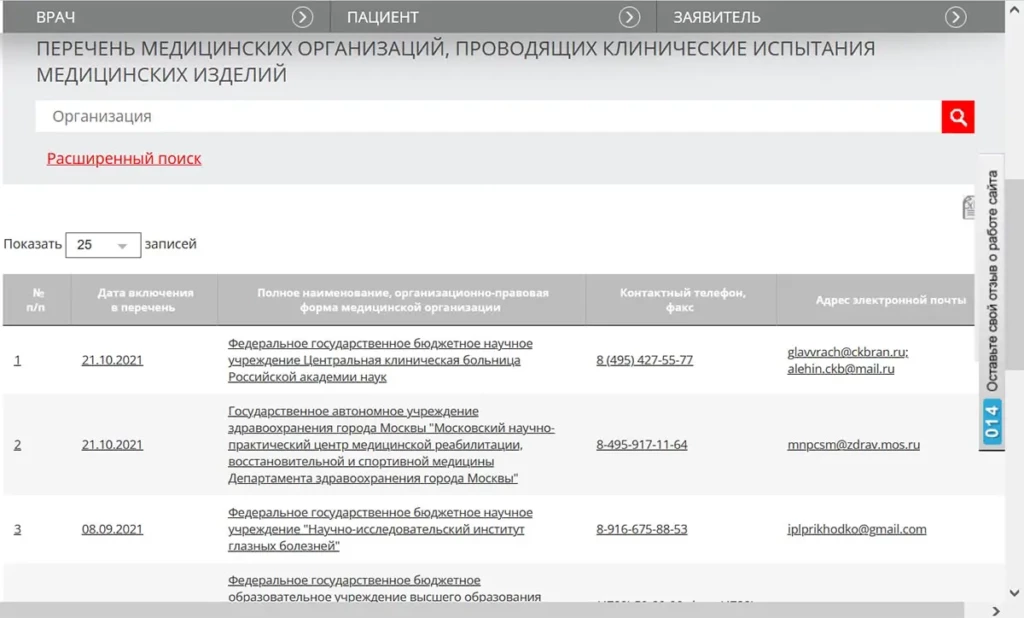
该电子服务包含以下信息
Этот электронный Сервис содержит сведения о:
机构列入名单的日期;
дате включения организации в Перечень;
医疗机构的名称和法律形式;
наименовании и организационно-правовой форме медицинской организации;
联系方式(电话、传真、电子邮件地址)。
контактах (телефоне, факсе, адресе электронной почты).
通过简单搜索,您可以根据机构名称找到该机构;通过高级搜索,您可以根据机构列入列表的日期找到该机构。
Простой поиск позволяет найти организацию по ее наименованию, а расширенный по дате включения в Перечень.
在选择医疗机构进行门诊时,必须考虑到该医疗机构的工作(服务)清单是否与许可证相符。例如,不能在没有妇科许可的医疗机构进行妇科椅试验。如果在没有合适许可领域的机构中进行临床试验,其结果将不被国家科研署接受。这可能导致产品注册被拒。
При выборе медицинской организации для проведения клиники необходимо учитывать её перечень работ (услуг) в соответствии с лицензией. Например, нельзя испытывать гинекологическое кресло в медицинском учреждении, в области лицензии которого отсутствует гинекология. В случае, если клинические испытания будут проведены в учреждении без подходящей области лицензирования, их результаты не будут приняты Росздравнадзором. Это может повлечь за собой отказ в регистрации изделия.
俄罗斯临床试验时间安排
Срок проведения
时间安排直接取决于所选择的形式。对于分析和评估现有数据的试验,平均时限不超过 1 个月。
Срок напрямую зависит от выбранной формы. Для испытаний в форме анализа и оценки доступных данных срок их проведения в среднем составляет не более 1 месяца.
如果需要进行以人为对象的临床试验,时间则取决于试验产品的类型。如果说通用诊断产品(超声波机、眼压计、密度计)的试验可以在 3-4 个月内完成,那么植入物(填充物、支架、心脏瓣膜假体)的试验则需要1年到数年的时间。
В случае если необходимо проведение клинических испытаний с участием человека, срок определяется видом испытываемого изделия. Если для диагностических изделий общего назначения (аппараты УЗИ, тонометры, денситометры) можно провести испытания за 3-4 месяца, то срок испытаний имплантатов (филлеры, стенты, протезы клапанов сердца) может составить от года до нескольких лет.
同时还应考虑到,只有在注册医疗器械的使用方面没有足够临床数据的情况下,才允许进行有人体参与的临床试验。因此,如果至少有一些临床数据,人体试验的范围和时间都可以大大缩短。
При этом необходимо учитывать, что допускается проводить клинические испытания с участием человека только по тем аспектам применения регистрируемого медицинского изделия, по которым отсутствуют в достаточном количестве клинические данные. Таким образом, при наличии хотя бы части клинических данных, объем и сроки испытаний с участием человека могут быть существенно сокращены.
临床和实验室试验(体外诊断产品)的时间通常在 1.5 至 4 个月之间。但是,如果需要采集罕见病理样本(例如,检测罕见遗传病的试剂及其试剂盒),时间可能会增加到 6-8 个月。
Срок проведения клинико-лабораторных испытаний (для изделий in vitro диагностики), как правило, составляет от 1,5 до 4 месяцев. Однако, в случае, если для них требуются образцы редких патологий (например, при испытаниях реагентов и их наборов для выявления редких генетических заболеваний), срок может увеличиться до 6-8 месяцев.
与其他类型的试验不同,临床试验不受第 885n 号命令规定的最长期限的限制!
В отличие от других видов испытаний, для клинических Приказом 885н не установлен максимальный срок их проведения!
我们在临床试验和医疗器械注册领域拥有多年经验,可以为您选择最合适的形式,并最大限度地缩短时间。
Многолетний опыт работы в сфере проведения клинических испытаний и регистрации медицинских изделий позволяет нам подобрать оптимально подходящую форму и максимально сократить их срок.
俄罗斯医疗器械临床试验费用
Стоимость клинических испытаний
要计算费用,您需要提供医疗器械注册档案的开发文件。
Для расчета стоимости необходимо предоставить разработанные документы регистрационного досье на Ваше медицинское изделие.
临床试验费用主要取决于医疗器械的以下参数:
Стоимость клинических испытаний в первую очередь зависит от следующих параметров медицинского изделия:
使用风险等级
класс риска применения;
用途
назначение;
新颖性和创新程度
новизна и степень инновационности;
是否有已注册的类似物;
наличие зарегистрированных аналогов;
是否有临床数据(临床前试验结果、临床报告、科学出版物、医生评论等)。
наличие клинических данных (результатов доклинических испытаний, клинических отчетов, научных публикаций, отзывов врачей и т.д.).
外,成本还取决于试验的形式:现有数据的分析和评估形式(无人员参与)、有人员参与的试验(研究)形式、临床和实验室试验形式(体外诊断产品)。
Кроме того, стоимость зависит от формы проведения испытаний: в форме анализа и оценки доступных данных (без участия человека), в форме испытаний (исследований) с участием человека, в форме клинико-лабораторных испытаний (изделия для in vitro диагностики).
因此,在对构成注册档案的现有文件进行详细研究后,可以计算出工作成本。
Таким образом, стоимость работ может быть рассчитана после подробного изучения имеющихся документов, составляющих регистрационное досье.
为什么选择我们?
上海经合俄罗斯专家自 2009 年以来一直从事医疗器械注册领域的工作。我们在注册和组织临床试验方面积累了丰富的经验。
公司专家在组织和开展有人体参与的多中心临床试验方面拥有成功经验,这些试验针对的是风险等级3类的产品,包括符合欧亚联盟要求的产品。
我们与被列入授权开展临床试验组织登记册的领先医疗机构建立了长期合作关系,确保我们能够提供最高性价比的服务,并避免在开展试验和处理结果方面出现延误。
我们不仅可以提供临床试验服务,还可以解决与获得注册证书有关的任何其他问题。如有必要,我们将根据试验期间收到的医疗机构建议,及时调整注册档案文件。
代替结论
对医疗器械进行临床试验是注册程序的关键部分。在这一阶段,要确定在医疗实践中使用该产品是否合理。
在试验过程中,医疗专业人员会根据积累的经验、现有的知识和提交的产品文件,对使用注册医疗器械可能带来的益处和可能造成的危害的比例得出结论。
我们公司专家深知临床试验阶段的重要性,愿意在与我们建立了长期富有成效合作关系的授权医疗机构提供交钥匙临床试验服务。我们将选择合适的试验形式,将提交试验的注册医疗器械样品数量减少到所需的最低限度,并确保在最短时间内以最佳成本获得试验结果。
如果您准备开展俄罗斯医疗器械注册,您可以通过任何便捷的方式联系我们,进行免费的初步咨询。
海关联盟EAC认证中心/上海经合工业设备检测有限公司
公司地址:上海浦东新区高科东路777弄1号楼2017室
联系我们:上海经合工业设备检测有限公司/俄罗斯EAC证机构中国代表处
电话:021-36411223 36411293
邮件:eac@cu-tr.org
手机微信:18621862553

欧亚联盟EAEU、俄罗斯、乌兹别克斯/哈萨克斯坦/白俄罗斯/乌克兰医疗器械和药品注册咨询服务
Eurasian Economic Union Russia Uzbekistan Kazakhstan Belarus Ukraine Medical Device and Drug Registration Consulting Services
Moscow, st. Lenin Sloboda, d.21, k.1 浙江省杭州市西湖区留和路129号2924室
China Office: Room 2017, Building 1, No. 777, Gaoke East Road, Pudong New Area, Shanghai,china
微信/电话:18621327282 QQ:594037022 gost-r@163.com 邮编:210201 海关编码查询 俄罗斯医疗器械注册证查询 俄罗斯医疗注册证书查询
网站内容和图片版权所有,未经允许,不得转载复制拷贝和使用 沪ICP备16045803号-6 沪ICP备10027014号-18 欧亚联盟药品注册证查询 俄罗斯计量注册证书查询
